|
La bêta-thalassémie est une maladie génétique qui touche l’hémoglobine, donc le transport de l’oxygène dans notre organisme. Dans ses formes les plus sévères, un suivi approprié et régulier permet d’en limiter les symptômes.  À quoi est due la thalassémie ?La bêta-thalassémie est due à une anomalie génétique héréditaire de l’hémoglobine.L’hémoglobine est composée de deux chaînes alpha et deux chaînes bêta. La fabrication des chaînes bêta est commandéepar les gènes « bêta globine ». Chaque individu possède deux gènes « béta globine ». L’un est transmis par le père, l’autre par la mère. Si les deux parents sont chacun porteur d’au moins un gène altéré, ils peuvent tous les deux transmettre ce gène altéré à leur enfant. La bêta-thalassémie correspond ainsi à la forme homozygote de la maladie : les deux gènes bêta transmis sont altérés ; par conséquent, les chaînes bêta sont produites en quantité insuffisante ou nulle. Ceci induit une production insuffisante d’hémoglobine globale. L’oxygène ne peut alors pas être transporté correctement dans le corps. La forme homozygote est ainsi souvent grave car elle se traduit par une anémie (un manque de globules rouges) très intense. Voici deux schémas vous permettant de comprendre le principe de transmission des gènes altérés En revanche, si un seul des gènes bêta transmis est altéré, on parle de forme hétérozygote. Celle-ci n’entraîne aucun symptôme gênant pour le patient mais peut être décelée sur une prise de sang car les globules rouges sont de très petite taille. 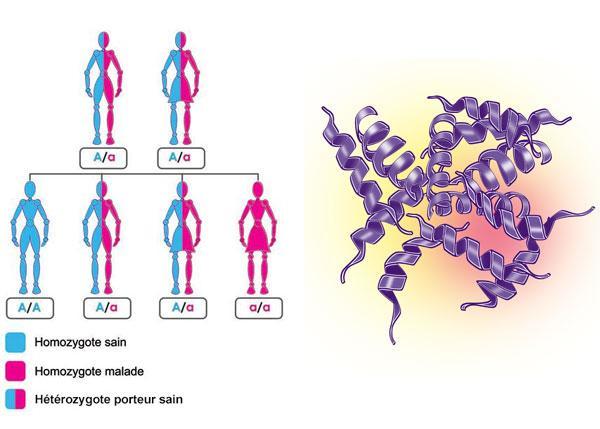 Qui peut en être atteint ?La bêta-thalassémie touche surtout les personnes originaires du bassin méditerranéen, du Moyen-Orient, d’Asie et d’Afrique noire, et autant les hommes que les femmes. On ne sait pas exactement combien de personnes en sont aujourd’hui atteintes. En France, environ 350 personnes présentent des formes sévères.Comment se manifeste la thalassémie ?Les symptômes sont plus ou moins précoces et plus ou moins sévères selon la production des chaînes bêta.La bêta-thalassémie majeure est aussi appelée anémie de Cooley. Les deux gènes bêta sont altérés (forme homozygote) et la production d’hémoglobine est très insuffisante voire nulle ; les symptômes sont sévères et apparaissent généralement entre 6 et 12 mois. Le nourrisson peut sembler fatigué, pâle et manger moins ; il peut parfois présenter une jaunisse (ictère). En cas d’anémie sévère prolongée, le volume du foie et de la rate augmente. Chez certains enfants, des manifestations osseuses peuvent apparaître. L’anémie sévère peut également conduire à un retard de croissance. La formation de calculs à l’intérieur de la vésicule biliaire peut également survenir. Ces calculs biliaires peuvent provoquer de fortes douleurs (en particulier la nuit ou après un repas). Il faut parfois retirer la vésicule biliaire par chirurgie. Les enfants atteints de bêta-thalassémie sévère, et dans une moindre mesure les adultes, sont très sensibles aux infections (surtout si leur rate a été enlevée), notamment au niveau des poumons, du cerveau et de l’organisme en général. Parfois, alors même que les deux gènes bêta sont touchés, les symptômes sont moins graves que dans l’anémie de Cooley. On dit qu’il s’agit d’une bêta-thalassémie intermédiaire. Les deux gènes bêta sont altérés, mais permettent la fabrication d’hémoglobine en petite quantité. La maladie se manifeste après l’âge de 2 ans. L’anémie est moins importante que dans la forme majeure. Les calculs biliaires sont fréquents. Les enfants atteints de cette forme de bêta-thalassémie ont une croissance normale, une puberté parfois retardée mais complète. L’augmentation du volume de la rate est très fréquente et est souvent une indication pour son ablation. Dans les bêta-thalassémies hétérozygotes , un seul des deux gènes bêta est altéré. En général, l’autre gène compense l’anomalie et produit suffisamment de chaînes bêta pour donner un taux d’hémoglobine normal ou proche de la normale. Les globules rouges sont en revanche plus petits quechez une personne bien portante. Comment détecte-t-on la thalassémie ?Le diagnostic de la bêta-thalassémie se fait sur simple analyse de sang quand un enfant présente des symptômes d’anémie. Le laboratoire d’analyse effectue une numération sanguine, qui consiste notamment à compter les différents éléments du sang, dont les globules rouges. C’est l’étude des différents types d’hémoglobine qui va permettre le diagnostic précis.Comment évolue la thalassémie ?Les formes mineures n’ont généralement aucune conséquence sur la santé.Les formes intermédiaires permettent aux enfants d’avoir une croissance et une puberté satisfaisantes. La bêta-thalassémie majeure ou anémie de Cooley nécessite la mise en place de transfusions régulières ou une greffe de moelle osseuse. Comment traite-t-on la thalassémie ?Le choix du traitement est influencé par l’âge du patient, la sévérité de la maladie et la réaction aux traitements. Dans les formes majeures, le traitement repose sur des transfusions sanguines régulières environ toutes les 3 semaines. Ceci est désormais possible grâce aux progrès de la transfusion sanguine mais malheureusement, la répétition des transfusions aboutit à surcharger l’organisme en fer. Lorsqu’il est présent dans l’organisme en trop grande quantité, le fer devient très toxique. C’est la raison pour laquelle il est essentiel de coupler les transfusions sanguines à un médicament susceptible d’éliminer le fer (on parle d’un médicament chélateur du fer). Longtemps uniquement disponibles en injection, les médicaments chélateurs du fer peuvent maintenant être souvent pris par voie orale.Enfin, la greffe de moelle osseuse permet de guérir la bêta-thalassémie, mais elle n’est actuellement pratiquée que dans environ un quart des cas car il faut qu’un frère, une sœur ou un parent soit compatible avec l’enfant au niveau de son type HLA. Quelle est la surveillance médicale dans le cas d’une thalassémie ?Les patients atteints de forme majeure reçoivent des transfusions environ toutes les 3 semaines. Un suivi médical régulier est pratiqué. Il permet de dépister d’éventuelles complications liées à la surcharge en fer et les effets secondaires des traitements.Mon quotidien va-t-il changer ?L’impact de la bêta-thalassémie sur la vie quotidienne dépend de la sévérité de la maladie.Le traitement est contraignant et doit être planifié : transfusions répétées, traitement chélateur. Néanmoins, lorsque le contrôle de la surcharge en fer est efficace, les personnes atteintes vont relativement bien et mènent une vie sociale proche de la normale, malgré parfois une tendance à se fatiguer plus vite que les autres ou une sensation de faiblesse récurrente. Pour les enfants, une scolarisation normale et la pratique d’activités sportives sont en général possibles. Il faut parfois établir un projet d’accueil individualisé (PAI) ou à un projet personnel de scolarisation (PPS). En cas d’hospitalisation, le suivi scolaire à domicile (service assistance pédagogique à domicile ou SAPAD) ou à l’hôpital est possible. Les adultes peuvent quant à eux exercer une activité professionnelle, aménagée ou non selon leur fatigabilité et la survenue d’éventuelles complications. Peut-on dépister la thalassémie avant qu’elle ne se déclare ?Si un des frères et sœurs est atteint, ou si les parents savent qu’ils sont porteurs de l’anomalie génétique, il est possible de réaliser un diagnostic prénatal à chaque grossesse. Son objectif est de déterminer au cours de la grossesse si l’enfant à naître sera malade ou non. Si le fœtus est atteint de la forme homozygote, une interruption de la grossesse sera généralement proposée.
Les commentaires sont fermés.
|
AuteurÉcrivez quelque chose à votre sujet. Pas besoin d'être fantaisiste, juste un aperçu. Archives
Juillet 2018
Catégories |
 Flux RSS
Flux RSS